


GIORNATA NAZIONALE PARKINSON 2024

In occasione della “GIORNATA NAZIONALE PARKINSON 2024” promossa dalla “Fondazione LIMPE per il Parkinson Onlus” con il patrocinio della “Società Italiana Parkinson e dei Disordini del Movimento LIMPE-DISMOV”
l’ambulatorio dei Disordini del Movimento della ASL Sassari partecipa con un evento informativo dal titolo “CORTES APERTAS”, che si svolge sabato 30 novembre dalle ore 09,30 alle ore 13,30 al Poliambulatorio ASL Sassari via Tempio 5.
A questa “Cortes Apertas” sono invitati le persone con un sospetto, oppure recente diagnosi, di malattia di Parkinson, che sinora non hanno avuto la possibilità di accedere all’ambulatorio dedicato della ASL Sassari.
Durante la mattinata si informeranno gli interessati ed i loro familiari sulle cause ed i fattori di rischio della malattia, e saranno illustrati i percorsi diagnostici e terapeutici utili e necessari per una corretta gestione globale della malattia di Parkinson.

A Tonino Marogna – testo di Franco Simula
A TONINO MAROGNA
Una folla di parenti, amici e soci dell’ Associazione Parkinson ha partecipato commossa al rito funebre di Tonino Marogna, celebrato mercoledì 30 ottobre 2024 nella chiesa della Sacra Famiglia.
Sin dalla fondazione dell’Associazione, Tonino è stato una colonna portante, dimostrando passione, dedizione, impegno e generosità.
Tonino Marogna è stato una roccia polimorfa, che è andato man mano sgretolandosi solo in questi ultimi mesi, ormai aggredito da un male incurabile, mai domo pur dopo aver superato un intervento chirurgico di notevole gravità.
Da sempre innamorato della moglie Adelaide, suo personale caregiver o come lui stesso scherzando si definiva, portatore sano di Parkinson, da sempre amico di Giuseppe Cossu col quale condivideva la passione per il calcio. Tonino fu dipendente attento al Petrolchimico di Porto Torres, dove teneva un occhio vigile alla salvaguardia del territorio dallo smaltimento delle scorie di lavorazione fino a quando, dopo l’esperienza turritana, venne assunto al Banco di Sardegna dove in breve divenne “la chiave” della cassaforte del Banco perché gli vennero affidate le mansioni di manutentore delle casse di sicurezza della Banca, ancora una volta come riconoscimento di dedizione e affidabilità.
Nella sua ormai non più breve storia l’Associazione Parkinson è stata ospitata in spazi diversi ma prevalentemente nelle scuole. Attualmente l’Associazione ha una sua sede propria, nella quale opera ormai da alcuni mesi e svolge le sue attività: ma lo spazio che Tonino ha vissuto, curato e frequentato più a lungo e con maggiore attenzione è stata la Casa di Via Rolando, avuta in affitto dall’Orfanotrofio. Lì ogni angolo reca la firma di Tonino, o per avere ripreso un pezzo di muro o per avere imbiancato o curato il giardinetto esterno, rifatti i serramenti, o per avere sempre, instancabilmente svolto il ruolo di custode e manutentore dei locali.
L’Associazione Parkinson è stata la passione più coinvolgente, da lui curata fin dalla fondazione originaria insieme ad Adelaide; grazie alla loro estesa rete di conoscenze sono riusciti a farci ottenere degli inaspettati benefici come ad esempio il Pullmino e svariate altre attrezzature d’ufficio.
Grazie,Tonino, vola verso le stelle nella luce eterna
Franco Simula
“HOUSTON, CI SENTITE?” di Kai S. Paulus

(Pillola n. 87)
Secondo il gruppo di ricercatori otorinolaringoiatrici statunitensi capitanati da Lee Neilson la riduzione dell’udito può rappresentare un fattore di rischio per sviluppare la malattia di Parkinson.
Whaoo, che frase forte!
Questa affermazione, pubblicata pochi giorni fa, il 21 ottobre scorso, è molto coraggiosa e provocatoria, visto che sinora si sosteneva che la perdita dell’udito non sarebbe correlato al rischio di riscontrare il Parkinson, come confermano anche il geriatra cinese Pingping Ning e colleghi solo pochi mesi fa. Poi, ieri, il 31 ottobre, il neurologo tedesco Thomas Mueller si inserisce nella discussione, ripercorrendo i dubbi su causa ed effetto, e cioè, se la neurodegenerazione comporti anche una compromissione dell’udito, oppure che i disturbi auricolari causino o comunque accelerino una neurodegenerazione.
Come vedete, botta e risposta in pochissimo tempo, il che sta a significare l’attualità della tematica nel contesto della corrente scientifica attuale che cerca indizi precoci per diagnosticare e trattare il Parkinson (vedi anche ” ALL ORIZZONTE LA DIAGNOSI PRECOCE DELLA MALATTIA DI PARKINSON”, “ PREVENIRE IL PARKINSON. PARTE 3: I PRODROMI“ e ” DIAGNOSI DI MALATTIA DI PARKINSON: CAMBIA TUTTO!“).
E’ davvero affasciante, perché stiamo assistendo, praticamente ‘in diretta’, ad una discussione scientifica internazionale di alto livello.
Ma di che cosa si tratta esattamente?
(da: Studio Sentire srl 2024)
Il nostro cervello si orienta elaborando continuamente segnali che provengono dall’ambiente circostante, e lo fa raccogliendo dati sensoriali (visivi, uditivi, tattili, termici, olfattivi) che poi servono per reagire, e determinano le nostre azioni ed i nostri comportamenti. Quando invece un canale sensoriale è difettoso e non arriva il solito flusso di informazioni, il cervello viene stimolato di meno e non svolge le abituali compiti correttamente; la ridotta stimolazione comporterebbe alla lunga una disfunzione dei circuiti cerebrali ed un maggiore rischio ad innescare oppure velocizzare dei processi degenerativi.
Ning e colleghi affermano di non aver riscontrato alcuna correlazione tra riduzione/perdita dell’udito e la malattia di Parkinson, mentre il gruppo di Neilson sostiene che la relazione esiste e che la correzione dell’udito con apparecchiature e protesi potrebbe ridurre il rischio di Parkinson.
Dal conto suo, Mueller prende atto del lavoro di Neilson ma non trova un chiaro collegamento tra disturbo otorino e malattia di Parkinson.
Continuiamo a seguire questa interessante discussione che aggiunge altri tasselli per una sempre migliore comprensione del funzionamento del cervello e del possibile sviluppo di patologie neurodegenerative.
Fonti bibliografiche:
Mueller T. Vorteil fuer Hoergeraetetraeger: Hoerverlust beguenstigt Morbus Parkinson. SpringerMedizin.de, Parkinson-Krankheit, Nachrichten, 31.10.2024.
Neilson LE, Reavis KM, Wiedrick J, Scott DS. Hearing Loss, Incident Parkinson’s Disease, and Treatment with Hearing Aids. JAMA Neurol, 21 october 2024; doi.org/10.1001/jamaneurol.2024.3568.
Ning P, Mu X, Guo X, Li R. Hearing loss is not associated with risk of Parkinson’s disease: a Mendelian randomization study. Heliyon, 6 June 2024; doi.org/10.1016/j.heliyon.2024.e32533.
DIAGNOSI DI MALATTIA DI PARKINSON: CAMBIA TUTTO! di Kai S. Paulus

(Pillola n. 86)
Sappiamo che la malattia di Parkinson è una malattia neurodegenerativa progressiva caratterizzata da sintomi motori, quali rallentamento motorio, rigidità muscolare, instabilità posturale e tremore a riposo, e da molti sintomi non motori, tra i quali ansia, depressione, disturbi del sonno, costipazione, riduzione dell’olfatto, dolori, fatica, e tanti altri.
Sappiamo anche che la diagnosi di Parkinson si basa sui sintomi cardinali (rallentamento, rigidità, tremore) e che non esistono né prevenzione e neanche alcuna terapia in grado di modificare il decorso della malattia.
Davvero?
No, in realtà le cose non stanno più così.
Da quest’anno, come raccontano le ricercatrici tedesche Henrike Knacke e Daniela Berg nell’attuale numero della rivista scientifica “InFo Neurologie+Psychiatrie“, la diagnosi della malattia di Parkinson è diventata molto più complessa, non più solo basata sull’osservazione clinica, ma ora include anche i reperti neuroradiologici (RM encefalo, SPECT cerebrali), le basi genetiche e la presenza o meno di alfa-sinucleina alterata nei liquidi corporei (saliva, liquor cefalorachidiano) e nella cute. E per la prima volta vengono considerati anche i sintomi precoci e preclinici, i cosiddetti “prodromi” (vedi anche “PREVENIRE IL PARKINSON. PARTE 3: I PRODROMI“), quali disturbi del sonno, costipazione, ipotensione ortostatica, disfunzioni erettili, disturbi funzionali vescicali.
Titolo dell’articolo di Henrike Knacke e daniela Berg “Diagnosi Parkinson: dalla classificazione clinica a quella biologica.” InFo Neurologie+Psychiatrie, ottobre 2024
Nel febbraio di quest’anno, il prof. Guenter Hoeglinger, insieme ad un prestigioso gruppo di esperti internazionali della malattia di Parkinson, ha proposto questo nuovo metodo di classificazione, SynNeurGe (che ricorda la parola inglese per “sinergia” e sta per Syn = alfa-sinucleina, Neur = neurodegenerazione, Ge = genetica) in cui vengono anche proposti per il periodo prima dell’esordio motorio della malattia tre fasi:
- Fase di rischio: condizionata dalla presenza di fattori genetici (predisposizione genetica), non modificabili, ed ambientali (esposizione a pesticidi, diserbanti, solventi, ecc.), modificabili (!!!)
- Fase preclinica: i processi neurodegenerativi sono iniziati ma ancora non rilevabili
- Fase prodromica: comparsa di sintomi non motori, non specifici, che però rappresentano un campanello d’allarme che permettono l’inizio dell’iter diagnostico e terapeutico, che rispetto a prima, può iniziare anche anni prima del noto esordio motorio del Parkinson.
Questo nuovo approccio diagnostico è molto importante per anticipare la diagnosi e migliorare la sua precisione, e soprattutto per sviluppare nuove strategie terapeutiche in grado di modificare il decorso della malattia, rallentarla, e per trovare terapie causali, cioè terapie per guarire dal rapace infingardo.
Il futuro è iniziato.
Fonti bibliografiche:
Hoeglinger GU, Adler CH, Berg D, Klein C, Outeiro TF, Poewe W, Postuma R, Stoessl AJ, Lang AE. A biological classification of Parkinson’s disease: the SynNeurGe research diagnostic criteria. Lancet Neurol, 2024;23(2): 191-204. doi: 10.1016/s1474-4422(23)00404-0.
Hoeglinger GU, Boxer AL, Lang AE. Clinical versus biomarker-based diagnosis of neurocognitive disorders. Lancet Neurol, 2024; 23(8): 765-766. doi: 10.1016/s1474-4422(24)00274-6.
Knacke H, Berg D. Diagnose Parkinson: Von klinischer zu biologischer Klassification. InFo Neurologie+Psychiatrie, 2024; 26(10): 40-49.
MISTER PARKINSON INCONTRA MISTER WILSON AD ALGHERO di Kai S. Paulus

Enorme quanto inaspettato è stato l’interesse per il primo incontro regionale sulla malattia di Wilson, che ieri, sabato 5 ottobre si è tenuto nella Sala Riunioni del Polisoccorso di Alghero.

La sala riunione del Polisoccorso Alghero ODV
L’idea, di parlare della malattia di Wilson, una rara malattia genetica (vedi ” ELEMENTARE, WILSON!”), hanno avuto il presidente della Associazione Parkinson Alghero ODV, Marco Balbina, ed il membro del Comitato Sardegna Associazione Nazionale Malattia di Wilson, Sara Rusconi. Si sono aggiunte subito altri esponenti del Comitato Sardegna, Marisa Cabiddu, Valeria Mulas e Donatella Secchi. Tutti insieme hanno organizzato un vero convegno scientifico con la partecipazione di esperto riconosciuti quali l’epatologo Giuliano Alagna (AOU Sassari), il pediatra Georgios Loudianos (Microcitemico Cagliari), il gastroenterologo e primario della Medicina Interna della ASL Nuoro Salvatore Zaru, insieme alle specializzande in gastroenterologia Laura Tanda e Donatella Dettori, lo psicoterapeuta Gian Luigi Pirovano (Alghero), e la psicologa Rossella Pinna (Sassari).
I presentatori Marco Balbina, Maria Antonietta Izza, Salvatore Dilorenzo, ed i rappresentanti del Comune di Alghero Francesco Marinaro e Enrico Bachisio Daga.
Dopo l’introduzione di Marco Balbina, della padrona di casa Maria Antonietta Izza (Polisoccorso Alghero ODV), che sarà anche la moderatrice dell’evento, e del presidente della Associazione Nazionale Malattia di Wilson Salvatore Dilorenzo, ed i saluti del Vice Sindaco del Comune di Alghero, Francesco Marinaro, e l’Assessore alla Programmazione, Bilancio e Risorse Umane del Comune di Alghero, Enrico Bachisio Daga, inizia una intensa ed interessantissima mattinata, che prima di iniziare vanta già curiosità e primati: la comunità del Parkinson invita quella del Wilson (personalmente lo trovo originale quanto azzeccato), è il primo convegno sulla malattia di Wilson in Sardegna, ed in sala sono presenti i massimi esperti regionali di questa patologia!

I relatori: Giuliano Alagna, Georgios Loudianos, Rossella Pinna, Laura Tanda, Donatella Dettori, Salvatore Zaru, Pier Luigi Pirovano.
Dalle relazioni dei sopracitati esperti è emerso che la malattia di Wilson, molto più frequente in Sardegna che non nelle altre regioni d’Italia, pur rimanendo una malattia fatale se non correttamente diagnostica e trattata, è gestibile e la terapia disponibile garantisce una aspettativa e qualità di vita pressoché sovrapponibile alla popolazione generale. Interessante è il fatto, che la malattia, che negli stadi avanzati colpisce il cervello causando un parkinsonismo, da molti anni in Sardegna non arriva più all’osservazione dei neurologi. Questo a dimostrazione dell’ottimo lavoro svolto da pediatri, epatologi e gastroenterologi nella precoce diagnosi e presa in carico di bambini ed adolescenti, un vanto della nostra sanità regionale che andrebbe maggiormente evidenziato.
La seconda parte della mattinata è stata dedicata alle struggenti testimonianze di mamme e ragazze.
In sala si fa silenzio quando Mariella prende la parola; parla di sua figlia Eleonora, del continuo peggioramento del suo stato di salute, l’odissea per tanti ambulatori e specialisti, quasi tutto a proprie spese (“quando tua figlia sta male non accetti che il CUP ti dia un appuntamento tra un anno”); Eleonora riceve tante diagnosi diverse, tranne quella che poteva salvarle la vita a 20 anni, ma la diagnosi di malattia di Wilson arriva troppo tardi. In sala si discute di errori umani e della gestione sanitaria, e tutti sono d’accordo, una storia così clamorosa noni deve ripetersi!

Le testimonianze di Emma, Viola, Mariella, Valeria, Donatella.
Emma, 14 anni, racconta la sua storia, del suo lungo malessere, fino a quando diagnosi e terapia le hanno conferito fiducia e la voglia di andare avanti, addirittura da voler diventare ricercatrice per studiare la sua malattia e poter aiutare altri ragazzi. Donatella, racconta la storia di suo figlio quindicenne Luca e come, dopo il lungo ed inspiegabile malessere del figlio, la diagnosi è stata quasi una liberazione e ritorno alla normalità.
Valeria invece parla del suo percorso che inizia all’età di sedici anni con vari disturbi cronici che solo dopo dieci anni, durante la gravidanza, trovano spiegazioni, ma con una costante e scrupolosa adesione alle prescrizioni mediche ha vissuto sinora per 30 anni una vita ‘normale’.
Infine, la giovanissima Viola presenta il libro “Progetto Luce” di Manuela Mirai, una favola illustrata che vuole aiutare i giovani pazienti a perdere la paura dalla malattia ed avere coraggio a vivere la loro vita, normale e piena di opportunità.

Altri momenti del convegno
Un convegno, pienamente riuscito: Mister Parkinson ha incontrato Mister Wilson, gli specialisti si sono confrontati con la realtà delle famiglie e delle persone ammalate, ed Alghero ha dato il via ad una discussione che sicuramente proseguirà con altre iniziative, con l’intento di informare e far conoscere una malattia rara, ma sottostimata, di offrire un punto di riferimento, e di togliere la paura, citando Salvatore Dilorenzo: “I draghi esistono. Ma come nelle fiabe si possono sconfiggere.”
MALATTIA DI HUNTINGTON (1) di Kai S. Paulus

(Pillola n. 83)
La malattia di Huntington è una rara patologia genetica dovuta ad una mutazione del gene della proteina huntingtina, HTT. Questa mutazione comporta una eccessiva ripetizione della tripletta di basi nucleotidiche (tutti i geni sono formati da una serie di nucleotidi), C-A-G, che, quando superano le 40 ripetizioni, porta alla formazione della proteina huntingtina alterata, tossica per la cellula nervosa.

Copertina del numero speciale sulla Malattia di Huntington della prestigiosa rivista scientifica “Movement Disorders” del novembre 2022.
Ma perché parlare della malattia di Huntington sul sito dedicato alla malattia di Parkinson?
- Entrambe le malattie sono delle patologie neurodegenerative progressive e basate sull’alterazione di una proteina essenziale per il corretto svolgimento delle funzioni dei neuroni,
- entrambe rientrano nel capitolo dei Disordini del movimento con interessamento del centro della selezione del movimento, i nuclei della base, al centro del cervello,
- l’Huntington viene utilizzato anche per studiare altre patologie neurodegenerative, come l’Alzheimer ed appunto il Parkinson.
Il fatto curioso è, che dal punto di vista clinico, una è il contrario dell’altra: il Parkinson è una malattia ipocinetica caratterizzata da rallentamento motorio, mentre l’Huntington è ipercinetico con eccesso di movimento.
QUADRO CLINICO
La “corea” viene descritta per la prima volta dal ventiduenne statunitense George Huntington nel 1872, ed è caratterizzata clinicamente dalla triade:
- Sintomi motori: le coree, cioè ampi movimenti involontari, irregolari, afinalistici, che inizialmente interessano un arto o una parte del corpo, ma che successivamente possono coinvolgere tutto il corpo; rigidità e distonie; perdita dell’equilibrio, disartria (difficoltà nell’espressione verbale), e disfagia (difficoltà a deglutire)
- Sintomi cognitivi: deficit della memoria a breve termine, disfunzioni esecutive (difficoltà nella pianificazione e riduzione delle flessibilità mentale), bradifrenia (rallentamento del pensiero)
- Sintomi psichiatrici: ansia, depressione, apatia, disturbo del controllo degli impulsi, aggressività, psicosi; tra le persone con Huntington il rischio di suicidio aumenta di sette-dodici (!) volte rispetto alla popolazione generale. Altro dato drammatico è l’ipersessualità all’inizio della malattia e l’iposessualità negli stadi più avanzati con ulteriore aggravamento della vita di coppia.
Tutti i sopramenzionati sintomi hanno un andamento progressivo iniziando lievemente ed aumentando nel corso della malattia diventando sempre più invalidanti, imponendo un crescente peso anche per la famiglia ed i caregiver.
Come nel Parkinson, l’esordio della malattia è preceduto da una fase prodromica, della durata anche di molti anni, in cui si presentano i futuri sintomi in forma molto lieve e che spesso sfuggono ad una diagnosi precoce.
Ma andiamo a vedere nella prossima parte che cosa succede esattamente nella cellula nervosa e perché e quando si sviluppa questa malattia.
(segue con “MALATTIA DI HUNTINGTON (2)“)
Tappe principali dei progressi nella ricerca sulla malattia di Huntington, con l’importante scoperta della mutazione genetica nel 1993 (da: Movement Disorders, 2022)
MALATTIA DI HUNTINGTON (2) di Kai S. Paulus

(Pillola n. 84, seguito di “MALATTIA DI HUNTINGTON (1)“)
Nella prima parte abbiamo conosciuto i sintomi ed il quadro clinico con cui si presenta questa malattia neurologica; ora vediamola un po’ più da vicino.
PATOGENESI
La causa della malattia è un’alterazione di una proteina, huntingtina, HHT, una grossa proteina che svolge diverse importanti funzioni dentro le cellule nervose: contribuisce alla struttura e stabilità cellulare come componente del citoscheletro, gioca un ruolo cruciale nello sviluppo del sistema nervoso centrale, ed è essenziale per il trasporto intracellulare di ‘mattoncini’ e trasmettitori (dopamina, ecc.), ma soprattutto l’huntingtina è coinvolta nella formazione e nel mantenimento della sinapsi, il punto di collegamento tra neuroni con cui viene trasmessa l’informazione nervosa.
Infine, e da non sottovalutare l’HTT serve per la sopravvivenza della cellula nervosa ed è coinvolta nei meccanismi che portano alla morte cellulare fisiologica e programmata, l’apoptosi (a differenza della morte cellulare da danno e malattia, la necrosi).
Ora, possiamo immaginarci cosa succede, quando questa importante componente cellulare non funziona correttamente.
E già, siamo alle solite; come sappiamo dalla alfa-sinucleina alterata, la proteina huntingtina mutata si aggrega e forma dei corpi di inclusione intracellulari.
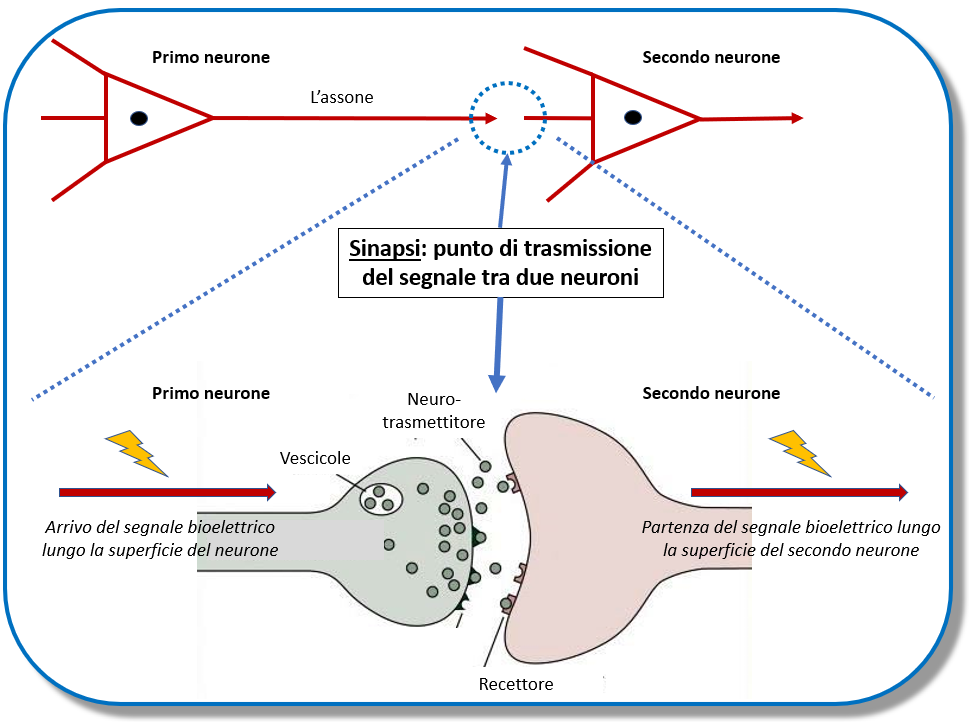
Un mio grafico che ho già proposto in altre occasioni, che vuole illustrare in forma semplificata la sinpasi, cioè il punto cruciale di contatto tra due neuroni, dove avviene la trasmissione dell’informazione neuronale
Vi ricordate i corpi di Lewy pieni di alfa-sinucleina? Ecco, nel caso della huntingtina mutata succede la stessa cosa: siccome gli aggregati di huntingtina mutata sono altamente dannosi per la cellula, quest’ultima si difende raccogliendo questi aggregati in dei sacchi di spazzatura pensandosi momentaneamente salva; ma la mondezza si accumula e finisce per occupare tutta la cellula che infine si deve arrendere. Però, questo è un processo relativamente lento. Ciò che è peggio è, che la proteina alterata non è più in grado di svolgere i propri compiti. E quindi la cellula nervosa avrà problemi strutturali e di stabilità, avrà problemi energetici per il danno provocato ai mitocondri, le centrali energetiche della cellula. Inoltre, il neurone non potrà garantire il trasporto ed il rilascio di neurotrasmettitori e la comunicazione tra i neuroni per l’alterazione delle sinapsi: e con la perdita di neurotrasmettitori e sinapsi la cellula nervosa perde il suo senso di esistere!
DIAGNOSI
La diagnosi di Huntington si basa sulla raccolta della storia personale e familiare, e sulla valutazione neurologica; Risonanza magnetica e/o TC cranio saranno utile per escludere altre problematiche neurologiche; con il test genetico si conferma infine il sospetto diagnostico.
TERAPIA FRAMACOLOGICA
A tutt’oggi non esiste una terapia farmacologica specifica per la malattia di Huntington, ma ci sono trattamenti sintomatici, come i cosiddetti antagonisti dopaminergici, utili a ridurre gli invalidanti movimenti coreici ma non privi di effetti collaterali, e farmaci dopaminergici, anti-parkinson, che aiutano in caso di rigidità e distonia.
Per le problematiche psichiatrici ci si avvale di ansiolitici, antidepressivi e neurolettici per mantenere il più a lungo possibile una sufficiente qualità di vita.
Detta così, la terapia sembra molto deludente, ma nell’ultima parte scopriamo i tanti interventi che si possono adottare per far stare meglio ammalati/e e familiari.
(segue con “MALATTIA DI HUNTINGTON (3)“)
Titolo della recentissima pubblicazione sugli attuali progressi nella ricerca della Malattia di Huntington, del gruppo di ricercatori cinesi intorno a Huichun Tong
MALATTIA DI HUNTINGTON (3) di Kai S. Paulus
(Pillola n. 85, seguito di “MALATTIA DI HUNTINGTON (2)“)
Nel capitolo precedente la terapia farmacologica ci ha delusi; in quest’ultima parte vediamo, che cosa c’é oltre le pastiglie.
TERAPIA NON FARMACOLOGICA
Ci sono tanti interventi a disposizione per garantire una corretta gestione globale delle persone. Vorrei iniziare con il supporto psicologico e psicoterapia: la persona ammalata percepisce il continuo peggioramento e la conseguente riduzione delle proprie autonomie, crescenti difficoltà personali, familiari e sociali, ci si sente soli ed incompresi. Ma anche i familiari necessitano di un sostegno psicologico, hanno bisogno di comprendere e di sapere come aiutare, e necessitano di suddividere le proprie energie per evitare un burn-out.
Essendo l’Huntington anche un disturbo del movimento, i trattamenti riabilitativi e fisioterapici dovrebbero iniziare sin dal momento della diagnosi, e la logoterapia appena si presentano difficoltà nel linguaggio e nella deglutizione.
STILE DI VITA
Anche qui troviamo analogie con il Parkinson: trattandosi di malattie neurodegenerative multifattoriali, anche se la genetica gioca un ruolo fondamentale nell’Huntington, dobbiamo cercare di limitare influenze e con-cause esterne ed ambientali.
- Dieta: tutte le malattie neurodegenerative comprendono dei meccanismi infiammatori sistemici e neurologici: una alimentazione sana ed equilibrata, con un intestino in salute riduce la neuro-infiammazione con frenata dei processi patologici. Il gruppo di ricercatori statunitensi intorno a Russel G. Wells propone frequenti digiuni per dare all’organismo la possibilità di depurarsi, riducendo la progressione della malattia, ed il gruppo austro-svizzero di Johannes Burtscher ricorda l’importante asse cervello-periferia per cui la correzione dello stile di vita e di alimentazione sono fondamentali per gestire l’Huntington.
- Attività fisica: essendo l’Huntington una malattia del movimento, è intuitivo che il movimento deve essere alla base di ogni intervento preventivo e di supporto nella sua gestione globale; quindi è bandito uno stile di vita sedentario.
- Sonno: il buon riposo notturno previene, ritarda e riduce ogni patologia neurodegenerativa, non per ultimo, per l’attivazione del sistema glinfatico, purificatore cerebrale, durante le fasi di sonno profondo.
PROSPETTIVE FUTURE:
La letteratura scientifica internazionale si sta arricchendo di continuo di nuovi studi e progetti per trovare cure efficaci per la malattia di Huntington. Si passa dalle immancabili cellule staminali, molto promettenti nei modelli sperimentali ma ancora difficoltose nell’uomo; dagli anticorpi (vaccinazioni) per eliminare gli aggregati di proteina alterata, ma bisogna risolvere il problema di far arrivare gli anticorpi a destinazione, troppo grandi per passare la barriera emato-encefalica; fino alla futura terapia genica, per riparare definitivamente il danno genetico.
IN CONCLUSIONE:
La malattia di Huntington è ancora una malattia fatale, ma, rispetto a solo pochi anni fa, oggi abbiamo a disposizione tanti strumenti per gestire meglio i sintomi, prolungare il periodo di sufficiente qualità di vita, ed interventi in supporto anche dei familiari e caregiver. Questo fa ben sperare che in questi anni arrivino importanti novità che possano rendere questa malattia molto meno terribile.
Fonti bibliografiche:
Burtscher J, Strasser B, Pepe G, Burtscher M, Kopp M, Di Pardo A, Maglione V, Khamoui AV. Brain-periphery interactions in Huntington’s disease: mediators and lifestyle interventions. International Journal of Molecular Sciences, 2024; 25: 4696. doi.org/10.3390/ijms25094696.
Kim KH, Song MK. Update of rehabilitation in Huntington’s disease: narrative review. Brain Neurorehabilitation, 2023;16(3): doi:10.12786/bn.2023.16e28.
Muehlbach A, Hoffmann R, Pozzi NG, Marziniak M, Brieger P, Dose M, Priller J. Psychiatrische Symptome der Huntington-Krankheit. Nervenarzt, 2024; 95: 871-884. doi.org/10.1007/s00115-024-01728-z.
Wells RG, Neilson LE, McHill AW, Hiller AL. Dietary fasting and time-restricted eating in Huntington’s disease: therapeutic potential and underlying mechanisms. Translational Neurodegeneration, 2024; 13(17): doi.org/10.1186/s40035-024-00406-z.
Tong H, Yang T, Xu S, Li X, Zhou G, Yang S, Yin S, Li XJ, Li S. Huntington’s Disease: Complex Pathogenesis and Therapeutics Strategies. International Journal of Molecular Sciences, 2024; 25, 3845. doi: 10.3390/ijms25073845.

Copertina di un attuale corso tedesco online sugli aspetti psicologici e psichiatrici della malattia di Huntington
PRIMA IL CERVELLO O PRIMA IL CORPO? di Kai S. Paulus

(Pillola n. 82)
Che cos’è la malattia di Parkinson?
Ma lo sappiamo tutti: un insieme variabile di sintomi motori, quali rallentamento e blocco motorio, rigidità, instabilità posturale e tremore. Giustissimo. E a causa di questi sintomi ci si reca dal neurologo specializzato in ‘disordini del movimento’.
Ma, come sappiamo, Parkinson è anche disturbo del sonno, depressione, dolori diffusi, riduzione dell’olfatto e disturbi intestinali, problemi spesso sottovalutati ed apparentemente non di pertinenza neurologica.
Disturbi intestinali? Ma il Parkinson non era una malattia neurologica?
Certamente, ma la sua origine non è necessariamente il cervello.
Se andiamo ad osservare la distribuzione della causa cellulare del Parkinson, l’alterazione della proteina strutturale e funzionale alfa-sinucleina (vedi ” L’ALFA-SINUCLEINA“), ci rendiamo conto che essa non si riscontra solo nel cervello, ma anche nella mucosa olfattivo del naso e nell’intestino, nel cuore e nella cute.
E c’è di più:
a seconda dove si accumula per prima la proteina alterata, nel cervello o perifericamente, si può distinguere una forma di Parkinson che origina nel cervello (“brain-first”, prima il cervello) ed un’altra che origina al di fuori di esso (“body-first”, prima il corpo), come raccontano gli studiosi danesi Jacob Horsager e Per Borghammer nel loro recente articolo sulla rivista scientifica “Parkinsonism and Related Disorders” (Parkinsonismi e malattie correlati).
Titolo della recente pubblicazione scientifica danese: “Prima-il-cervello versus prima-il-corpo di malattia di Parkinson: un aggiornamento su recenti evidenze”.
Queste due forme di malattia di Parkinson si distinguono, secondo gli autori scandinavi, non solo per distribuzione di alterata proteina, ma anche per sintomi e quadro neurologico.
E quindi:
La forma “prima il cervello” sarebbe caratterizzata da sintomi motori (rallentamento, rigidità, instabilità) e tremore prevalente ad un lato del corpo, con solo occasionale stitichezza, raro disturbo comportamentale del sonno REM, olfatto quasi normale, e buone condizioni cognitive.
Invece, la forma “prima il corpo” si distinguerebbe per la presenza di sintomi motori meno asimmetrici e maggiore instabilità posturale, chiaro disturbo comportamentale del sonno REM, frequente stitichezza, frequente ipotensione ortostatica (calo pressione quando si sta in piedi), e iniziali problemi cognitivi.
I danesi basano la loro distinzione del Parkinson in due entità in base al riscontro di accumulo di alfa-sinucleina nei nuclei della base dentro il cervello nel caso della forma “prima il cervello”, invece nell’intestino, nel cuore e nella cute. In entrambe le forme possiamo trovare proteine alterate nella mucosa olfattiva.
Concludendo:
Da queste osservazioni segue, che la forma di Parkinson “prima il cervello” sia dovuta principalmente a predisposizioni e mutazioni genetiche che riguardano le strutture motorie del cervello, mentre la forma di Parkinson “periferico” possa essere ricondotta prevalentemente a cause ambientali (vedi “BUON MICROBIOTA = MENO PARKINSON“).
La conferma di questa suddivisione del nostro rapace infingardo avrà degli importanti risvolti pratici, di cure e di prevenzioni, di cui parleremo sicuramente prossimamente.
Fonte bibliografica:
Horsager J, Borghammer P. Brain-first vs body-first Parkinson’s disease: an update on recent evidence. Parkinsonism and Related Disorders, 2024; 122: 106101. doi: 10.1016/j.paerkreldis.2024.106101.